Home
About
Journal Overview
Indexed
Copyright Policy
Policy on The Sharing of Paper Related Data
Contact Us
Information for Authors
Scope and Paper Type
Basic Requirements for Manuscript
Submission Guidelines and Important Notes
Review Policy
Publishing Process
Correction and Retraction
Requirements for Revised Manuscript
References Format
Publication Charges
Subscription
Editorial Board
Present Editorial Board
Youth Editorial Board
Download Centre
中文
Differential Role of Rotational Positioning in Pioneer Transcription Factor Binding to Nucleosomes
In vivo vs. In vitro
The Mechanism of Conformational Transition and Stabilisation Strategies of Viral Membrane Fusion Proteins
Implementing The IPDPS Teaching Concept in“ Biology in Daily Life” General Education Course for Cultivating Elite Innovators at Universities
Research on Multi-dimensional Feature Fusion Model for Osteoporosis Risk Assessment Based on Deep Learning
Treg Cells and Peripheral Immune Tolerance: From Discovery to Precise Immune Regulation
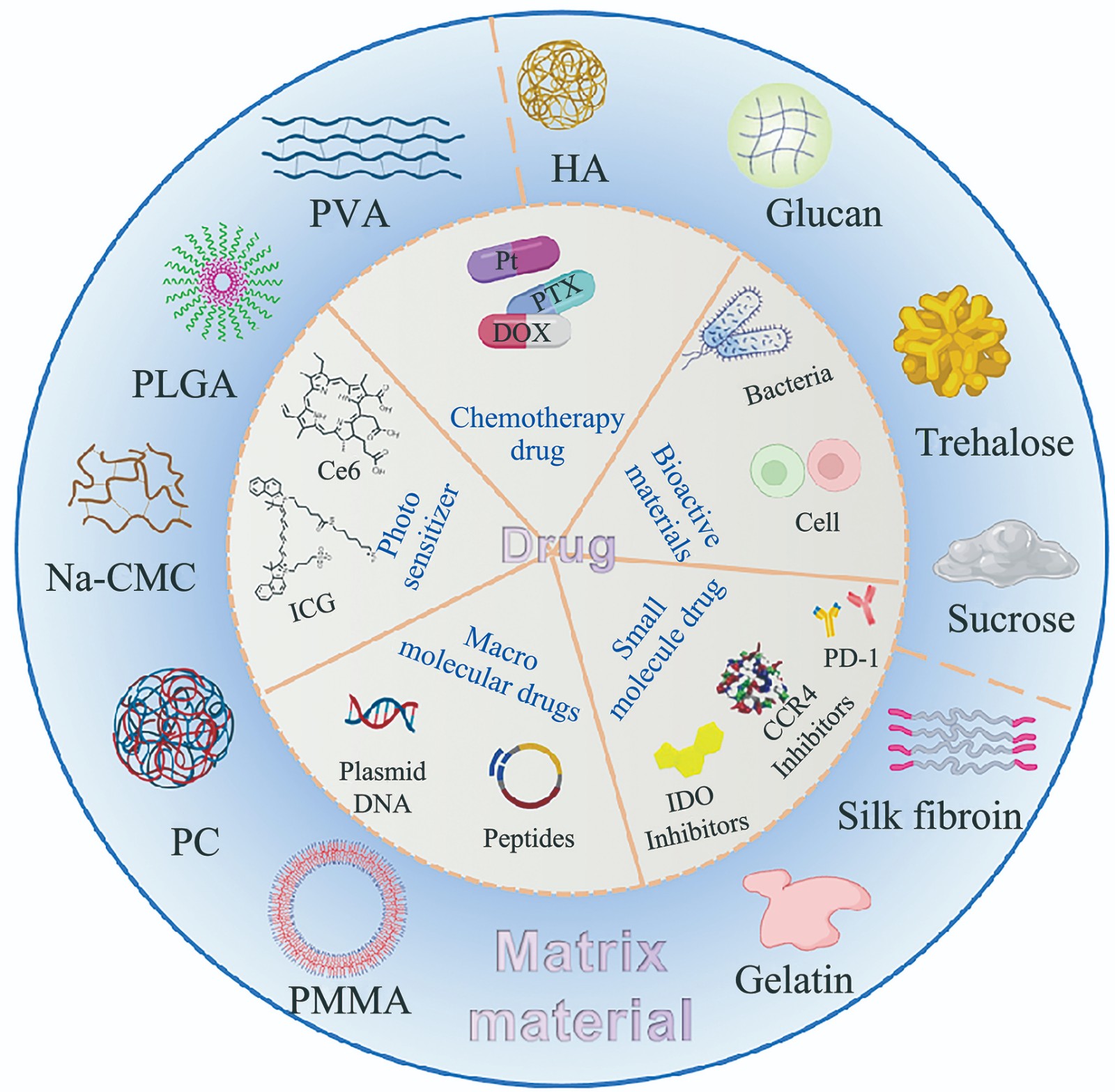
From Self-assembly to Smart Delivery: Construction Strategies and Frontier Applications of Prolamin-based Multicomponent Complex Nanocarriers
Discovery of Regulatory T Cells and Their Prospective Therapeutic Applications
Development and Application of Proximal Biotin Labeling Techniques
The Cellular Mechanism of Irisin in improving Diabetic Cardiomyopathy
Research on Microwave Induced Thermoacoustic and Ultrasound Dual-modality Microscopy
Artificial Intelligence for Nucleic Acid Aptamers: Methods and Applications
Multidimensional System of Precision Exercise Interventions for Parkinson’s Disease: Dynamic Regulation Based on Genetic Typing, Motor Subtypes, Clinical Staging, and Wearable Digital Biomarkers
Network Pharmacology and Experimental Verification Unraveled The Mechanism of Pachymic Acid in The Treatment of Neuroblastoma
The Critical Roles of GABAergic Interneurons in The Pathological Progression of Alzheimer’s Disease
Analysis of T7 RNA Polymerase: From Structure-function Relationship to dsRNA Challenge and Biotechnological Applications
Single-cell Protein Localization Method Based on Class Perception Graph Convolutional Network
Tumor Microenvironment Polyamines Inhibit T Cell Antitumor Activity
The Role of Golgi Apparatus Homeostasis in Regulating Cell Death and Major Diseases
The Regulatory Mechanisms of Dopamine Homeostasis in Behavioral Functions Under Microgravity
Junctophilin-2 MORN-Helix Domain: Structural Basis for Membrane Binding and Hypertrophic Cardiomyopathy-associated Mutations
PES1 Repression Triggers Ribosomal Biogenesis Impairment and Cellular Senescence Through p53 Pathway Activation
Application of Nanomaterials in The Prevention and Treatment of Radiation-induced Injury
Mitochondrial Function and Regulation in Spermatogenesis and Activation of
Caenorhabditis elegans
Adhesion Mechanisms of Aquatic Fouling Organisms Mediated by Biomacromolecules
The Mechanisms of Neurotransmitters and Their Receptors in Exercise Central Fatigue
The Mechanism of Exercise Regulating Intestinal Flora in The Prevention and Treatment of Depression
Regulation of Immune Function by Exercise-induced Metabolic Remodeling
Effects of Exercise Training on The Behaviors and HPA Axis in Autism Spectrum Disorder Rats Through The Gut Microbiota
Intergenerational Effects on Metabolic Health: Perspectives on Maternal Nutrition and Exercise During Pregnancy
Neuroplasticity Mechanisms of Exercise-induced Brain Protection
Research on The Construction and Application of Multiple Fluorescence Amplification System for Three Kinds of Stains
Inhibition of HDAC3 Promotes Psoriasis Development in Mice Through Regulating Th17
Applications of EEG Biomarkers in The Assessment of Disorders of Consciousness
Therapeutic Study on The Inhibition of Neuroinflammation in Ischemic Stroke by Induced Regulatory T Cells
The Current Status of Research on The Association Between
TMEM43
Gene and Hearing Loss
Role of SPINK in Dermatologic Diseases and Potential Therapeutic Targets
Translational Research of Electromagnetic Fields on Diseases Related With Bone Remodeling: Review and Prospects
Optimization of Ovarian Tissue Vitrification Using Hydrogel Encapsulation and Magnetic Induction Nanowarming
Wdr63
Deletion Aggravates Ulcerative Colitis Likely by Affecting Th17/Treg Balance and Gut Microbiota
Applications of Vaterite in Drug Loading and Controlled Release
Role of Innate Trained Immunity in Diseases
High Expression of INF2 Predicts Poor Prognosis and Promotes Hepatocellular Carcinoma Progression
Insights on Peripheral Blood Biomarkers for Parkinson’s Disease
Nucleic Acid-driven Protein Degradation: Frontiers of Lysosomal Targeted Degradation Technology
The Discovery of microRNA and Its Significance: The Enlightenment of The Nobel Prize in Physiology or Medicine of 2024
Intelligent Protein Engineering
Research Progress and Biomedical Applications of Magneto-controlled Nanobiocatalysis
CDK8 Promotes Cell Proliferation, Migration and Invasion in Esophageal Squamous Cell Carcinoma Through JAK/ STAT3/EMT Pathway
Exercise Improves Nonalcoholic Fatty Liver Disease in T2DM Mice by Inhibiting Ferroptosis Through p38 MAPK Signaling Pathway
Functions of Dynamin and Its Family Proteins
Methods for Inducing Homologous Protein Dimerization
Application of Functionalized Liposomes in The Delivery of Natural Products
Nanozyme and Abiogenesis
Wnt/β-catenin Signaling Cascades in Cardiovascular Diseases
The Regulatory Role of microRNA in Neocortical Layer Formation
The Neural Network Representation of Pain in Humans
Mitochondrial Regulation of Tumor-associated Macrophages
Immunotherapy for Colorectal Cancer
Advances of Volume Electron Microscopy
Frontiers in in situ Cryo-electron Microscopy and Visual Proteomics
Metabolomic Analysis of Mesenteric Lymph Fluid in Rats After Alcohol Gavage
Full-field Anterior Chamber Angle Measurement Based on Optical Reflection Tomography
Development and Application of Detection Methods for Capture and Transcription Elongation Rate of Bacterial Nascent RNA
The Magnetic Field Effects on The Oxygenation Rate of Recombinant Hemoglobin
Detection of Water Distribution in Plant Leaves Using Thermoacoustic Imaging
Study on The Promotion of Tenocyte Proliferation and Differentiation by Oriented Fiber Membrane Loaded With Nano-zinc Oxide
Application and Prospects of Polygenic Risk Score( PRS) in Genetic Disease Research: a Review of Data Analysis Methods
Screening and Functional Analysis of BACE1 Interacting Proteins in Alzheimer’s Disease
Vitamin D Plays a Crucial Role in Regulating Dopamine Nervous System in Brain
A Multiplex Network Control Method for Identifying Personalized Cancer Driver Genes
Effect of Combined Frequency Stimulation on The Electrophysiology of Granule Neurons in The Hippocampal Dentate Gyrus Area of Hindlimb Unloading Mice
Nanomaterial-based Therapeutics for Biofilm-generated Bacterial Infections
Ku70 Functions as an RNA Helicase to Regulate miR-124 Maturation and Neuronal Cell Differentiation
The Functional Role of SUMOylation in The Tumor Microenvironment
PRMT7 Regulates Adipogenic Differentiation of hBMSCs by Modulating IGF-1 Signaling
The Role of Ubiquitination in Regulating Ferroptosis
Neural Representation of Multiple Spatial Scales
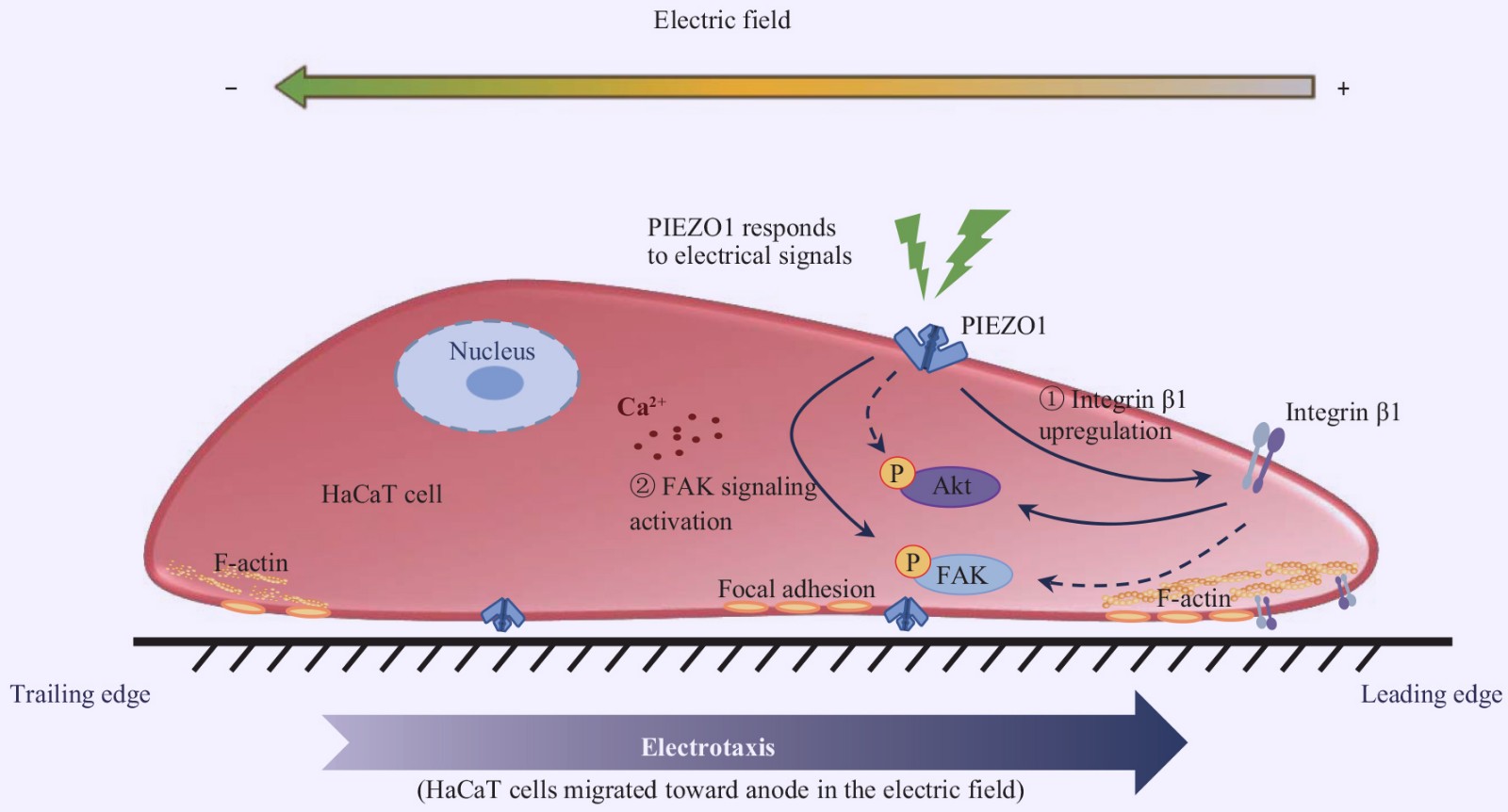
PIEZO1 Channel is Involved in Electric Field Guided Cell Migration
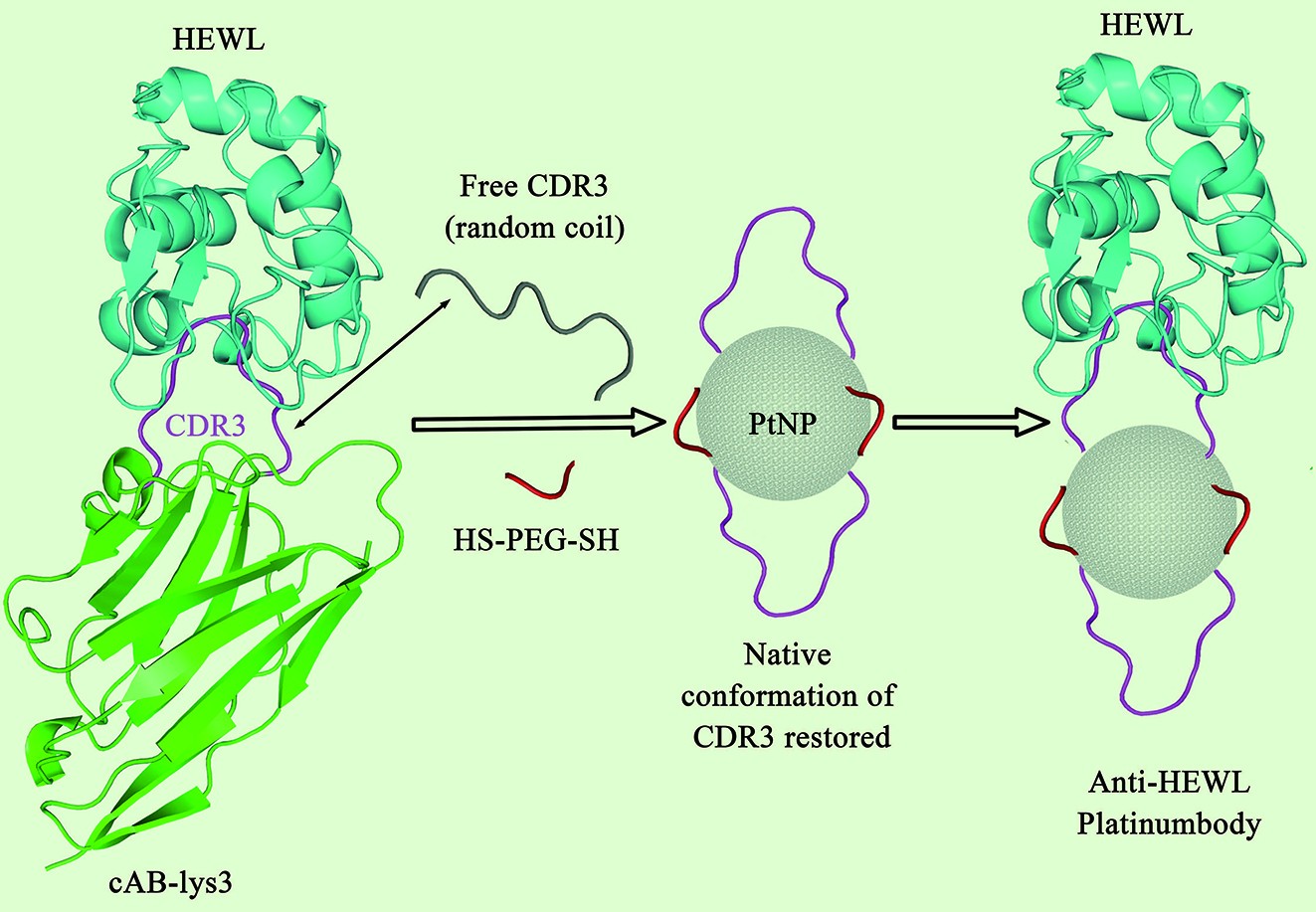
Conformational Engineering of Antibody Fragments on The Surface of Platinum Nanoparticles
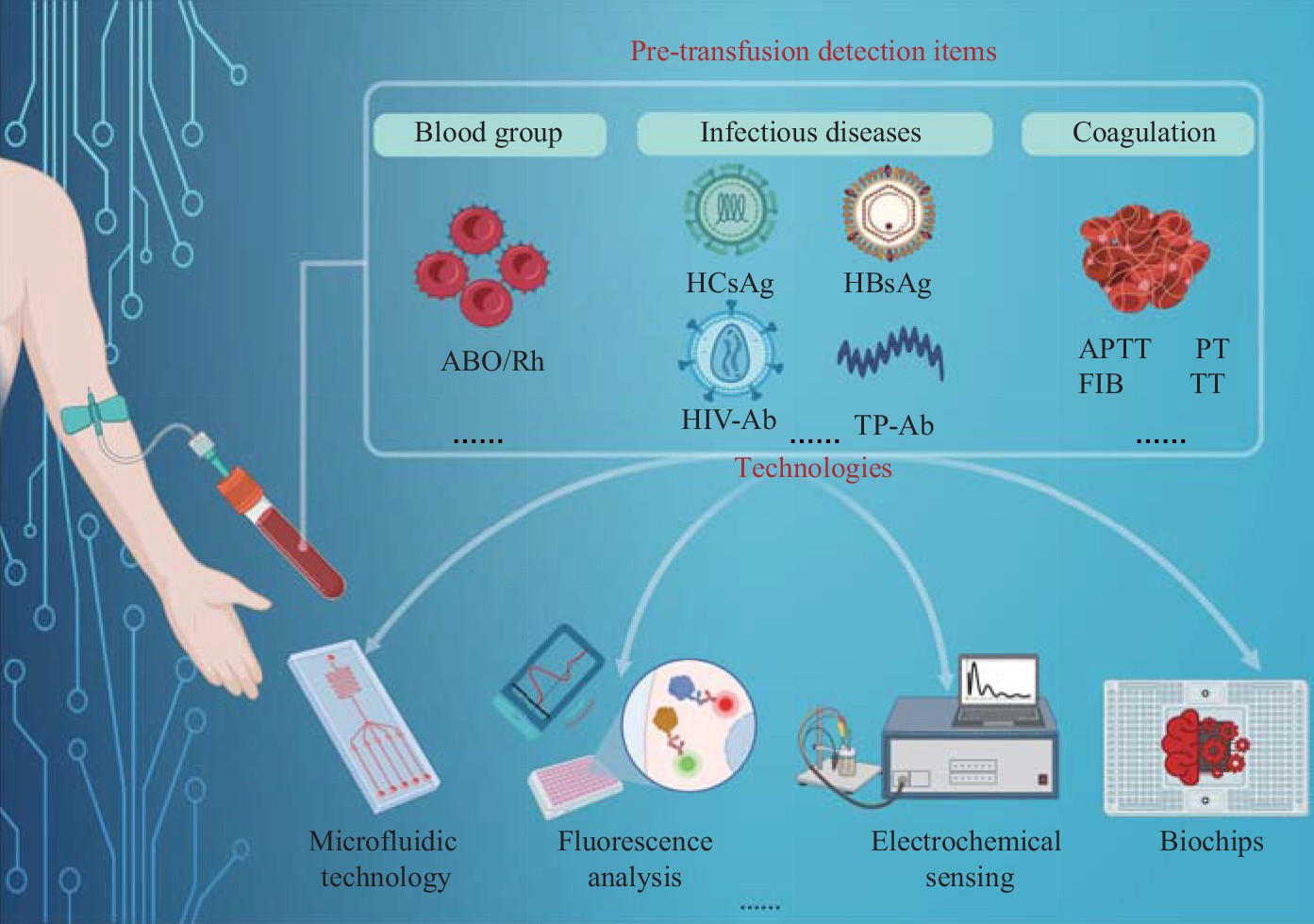
Application and Prospect of Pre-transfusion Detection Technology
Polycystin-2 Ion Channel Function and Pathogenesis in Autosomal Dominant Polycystic Kidney
Physicochemical Processes of Biofilm Formation on The Surface of Structures in Water
Extracellular Acidification Impairs Macrophage Lipophagy Through ASIC1/RIP1 Pathway
Research on Automatic Microalgae Detection System Based on Deep Learning
Biocompatible Transdermal Microneedles for Superficial Tumor Therapy
Design and Enzyme-mimicking Activity of High-loading Cu Single-atom Nanozyme
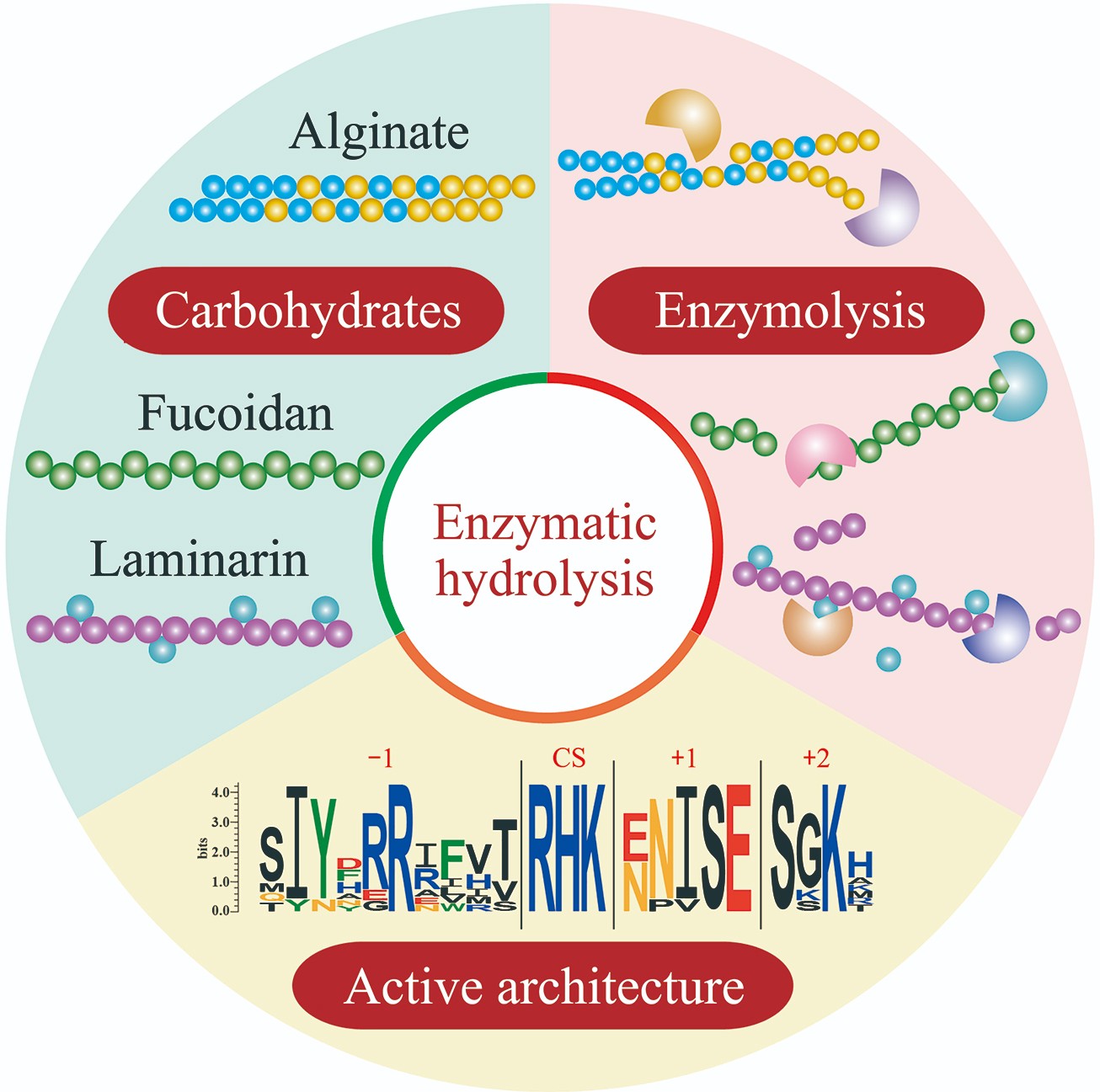
Complex Structure, Degrading Enzymes and Biological Activity of Brown Algae-associated Polysaccharides
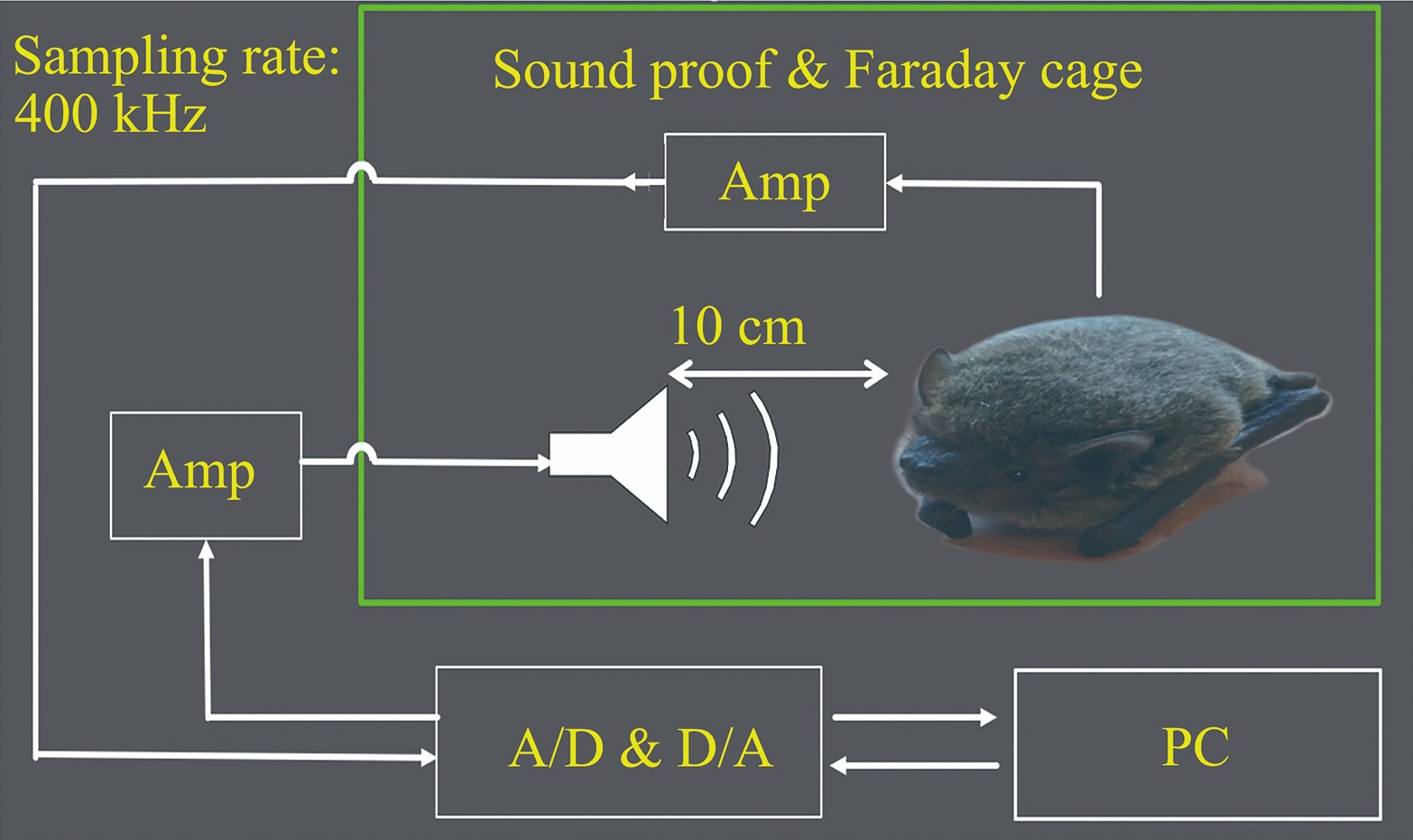
Special Frequency Suppression in Frequency Modulation Bats Reflected in Cochlear Microphonics
The Application of Neuromodulation Technology in Cognitive Impairment of Alzheimer’s Disease
The Mechanism of Exercise Regulating The Autophagy-lysosome Pathway to Prevent Alzheimer’s Disease
Efficacy of Tai Chi Exercise With Different Training Loads on The Rehabilitation of Patients With Parkinson’s Disease in The Early and Middle Stage
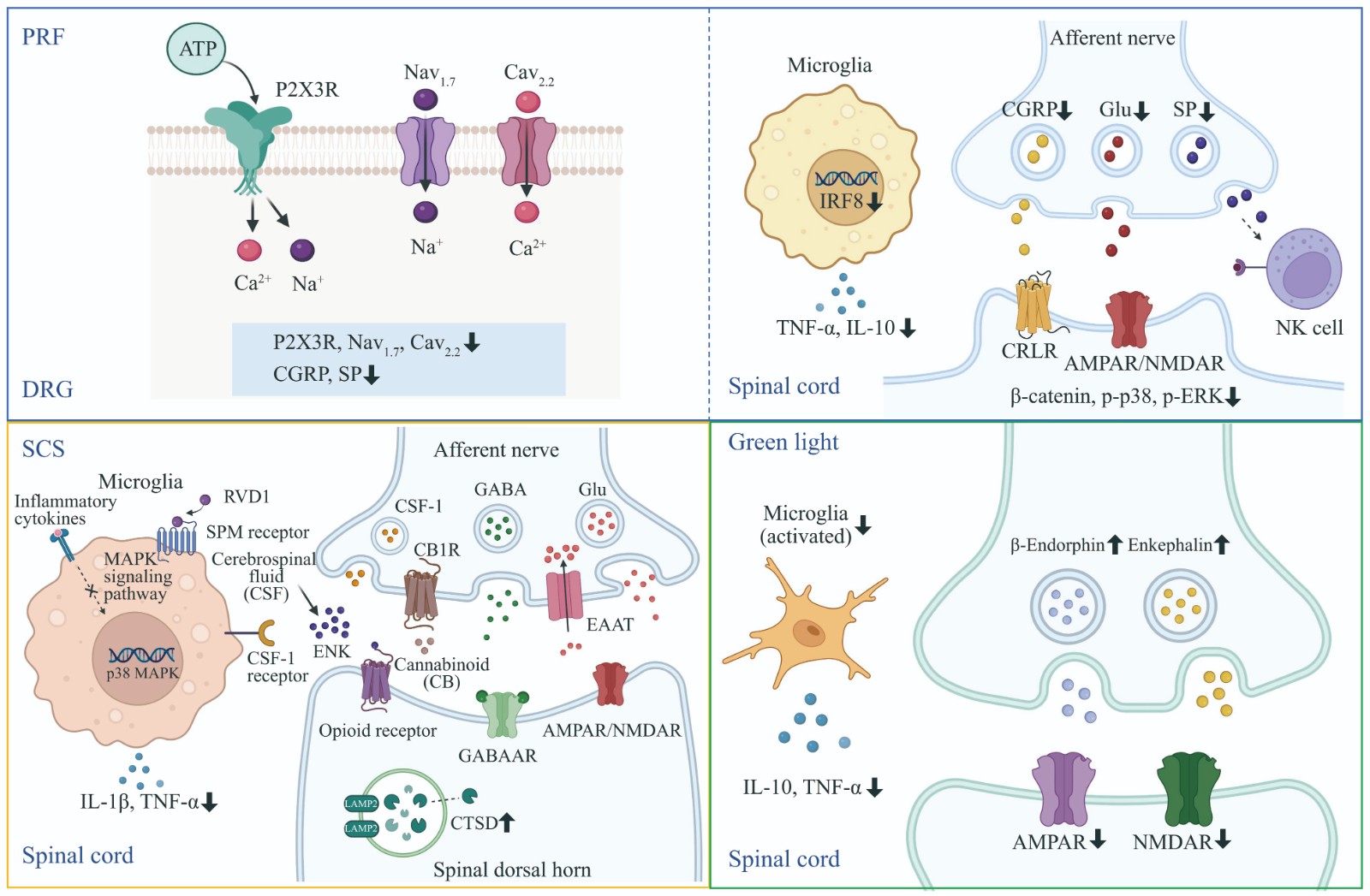
Application and Therapeutic Mechanisms of Non-pharmacological Interventions in The Management of Chronic Pain
Anti-tumor Study of DNA-RNA Nanocarriers Linked to AS1411 Aptamer
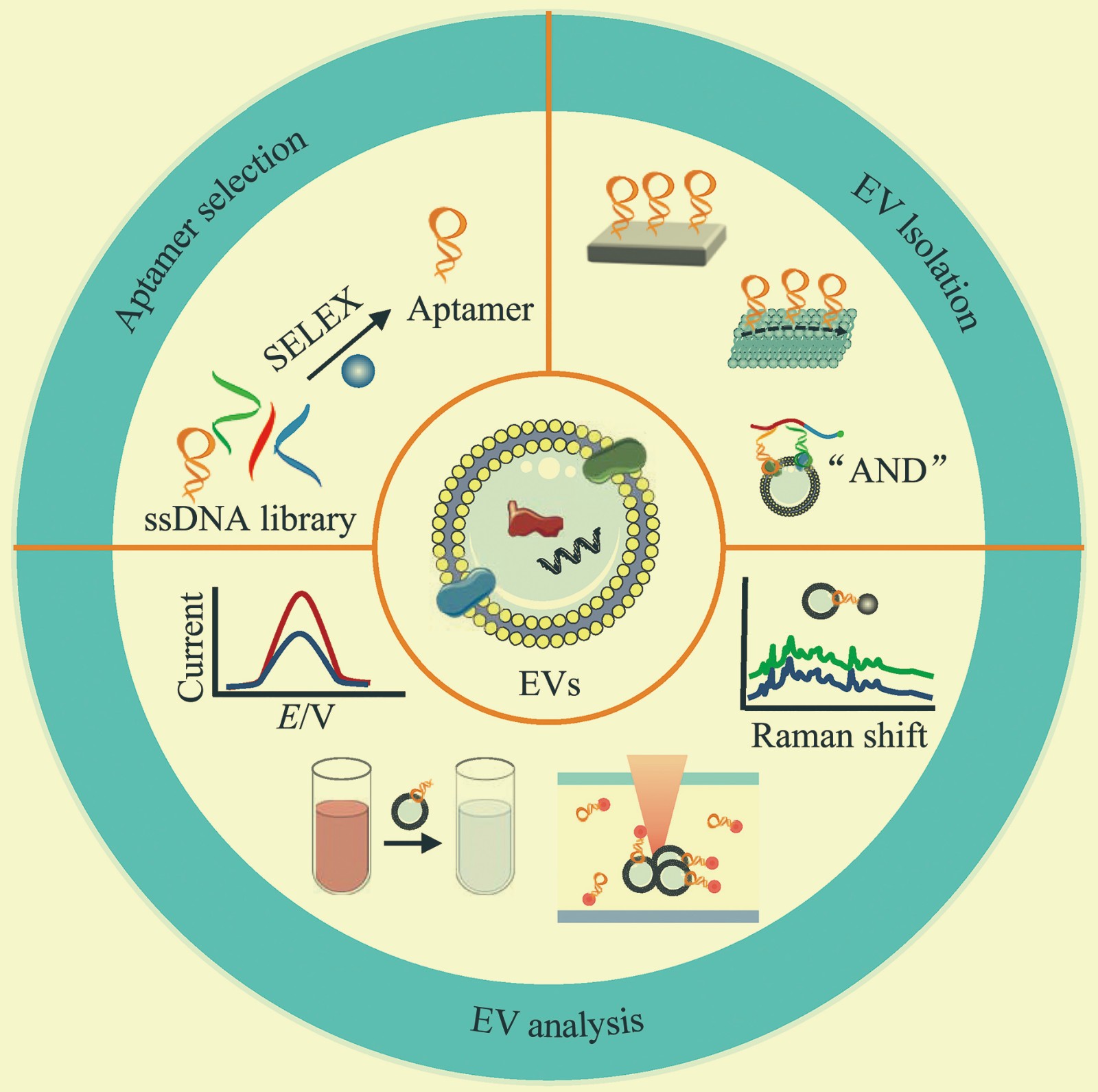
Aptamer-based Isolation and Analysis Methods for Extracellular Vesicles
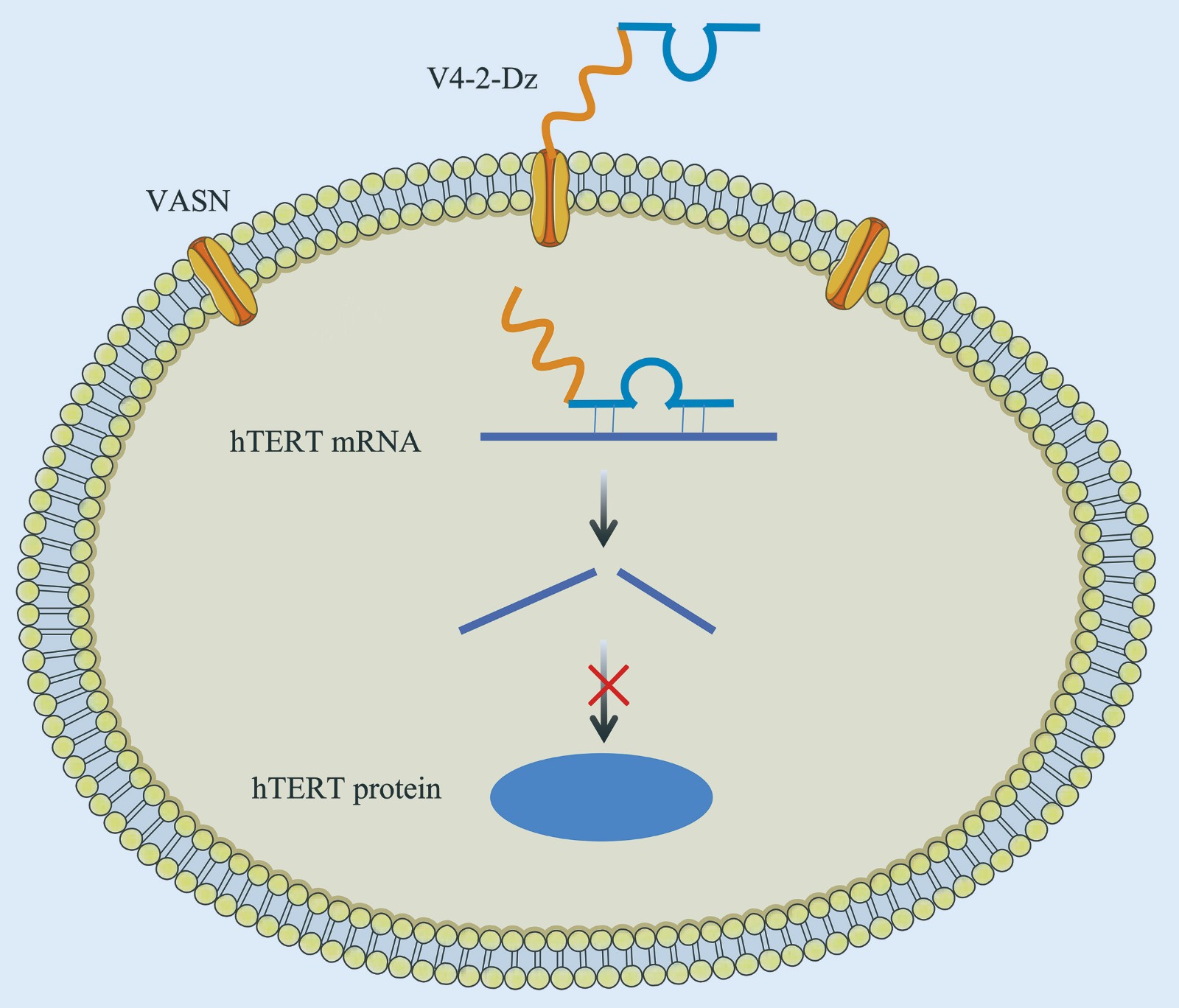
Functional Study of Conjugate of VASN Aptamer-DNAzyme for Telomerase Reverse Transcriptase
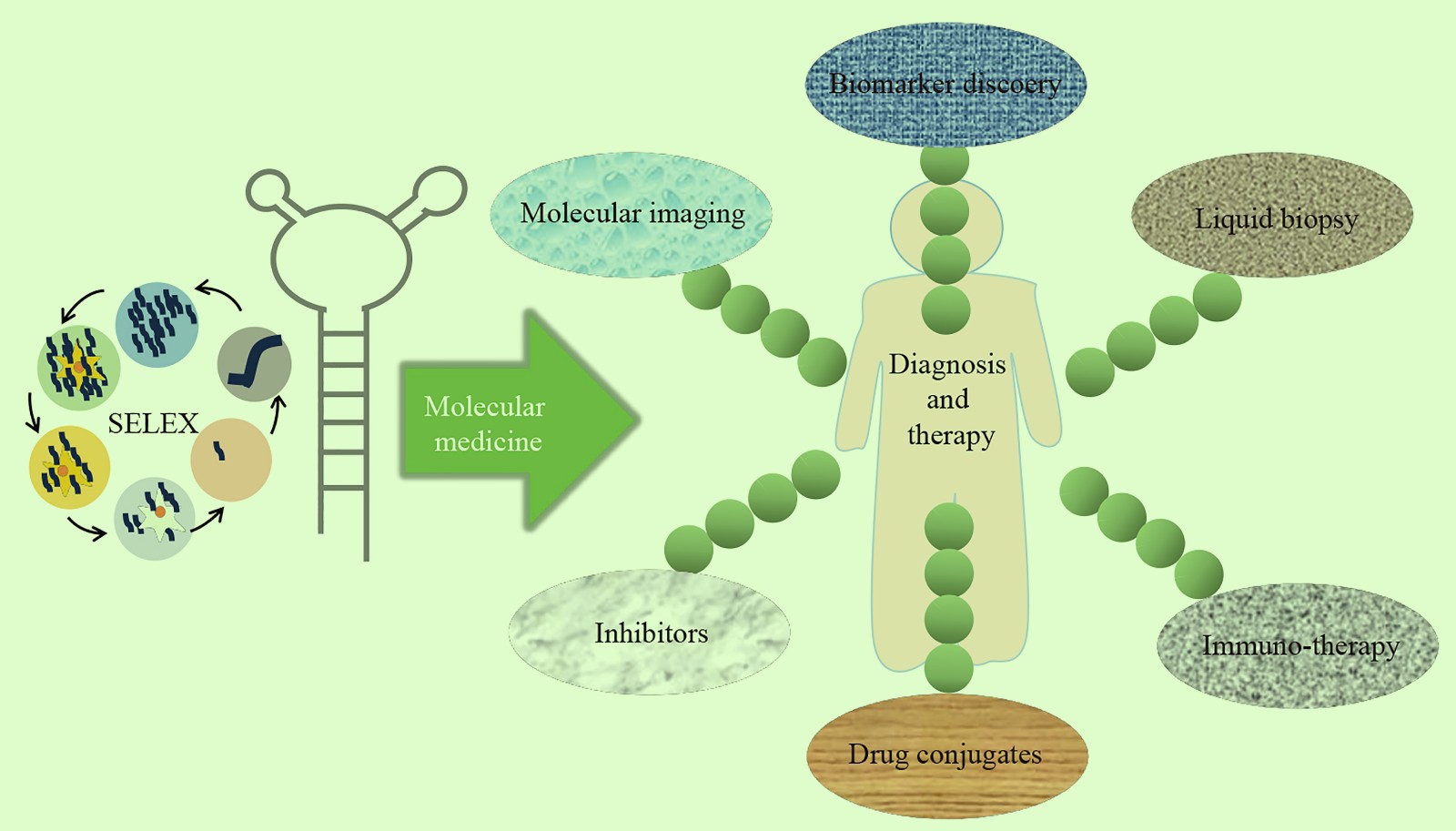
Applications of Aptamers in Molecular Medicine
LBP1C Extracted From
Lycium barbarum
Delays Aging by Activating TFEB
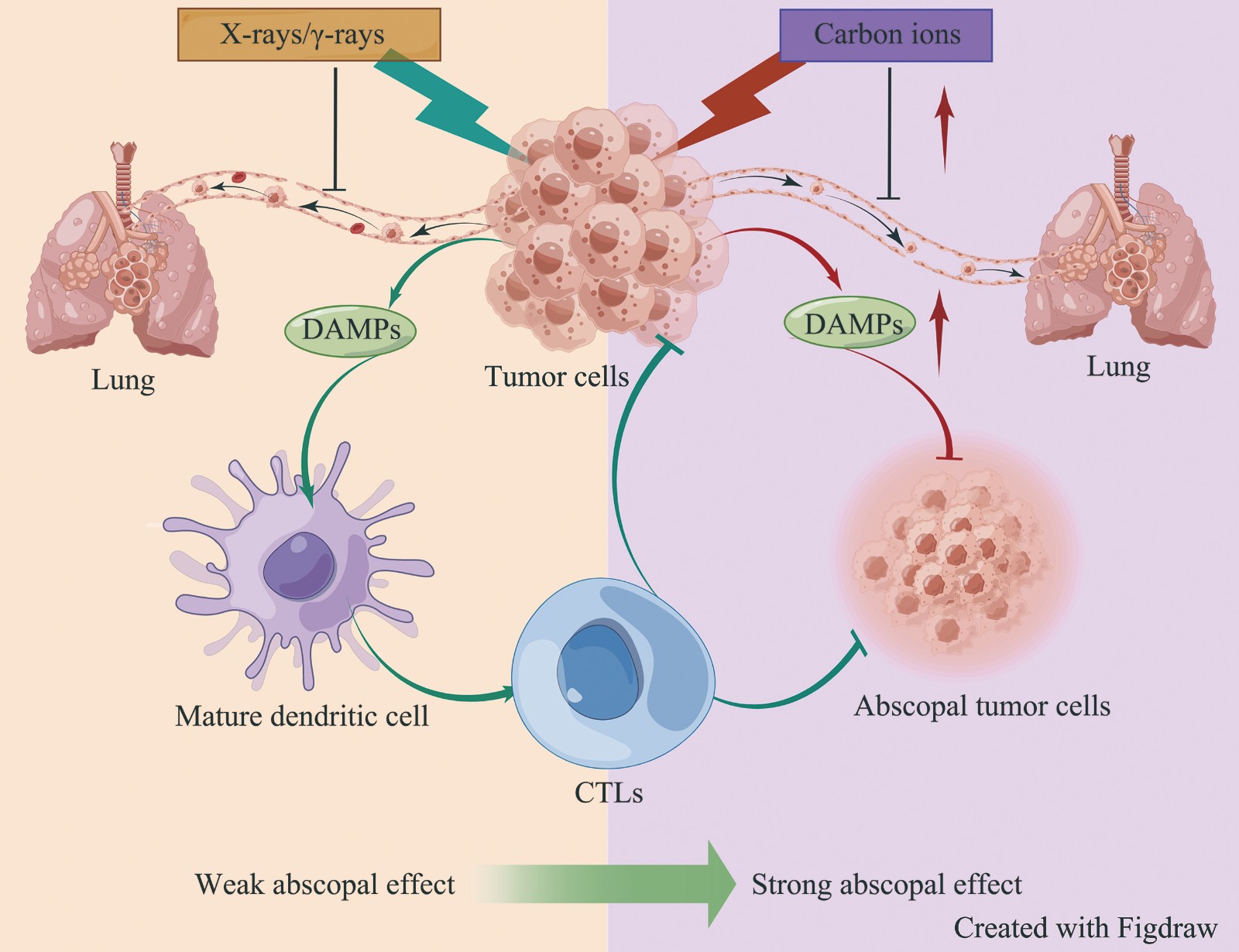
Abscopal Effect Induced by Carbon Ions Radiation
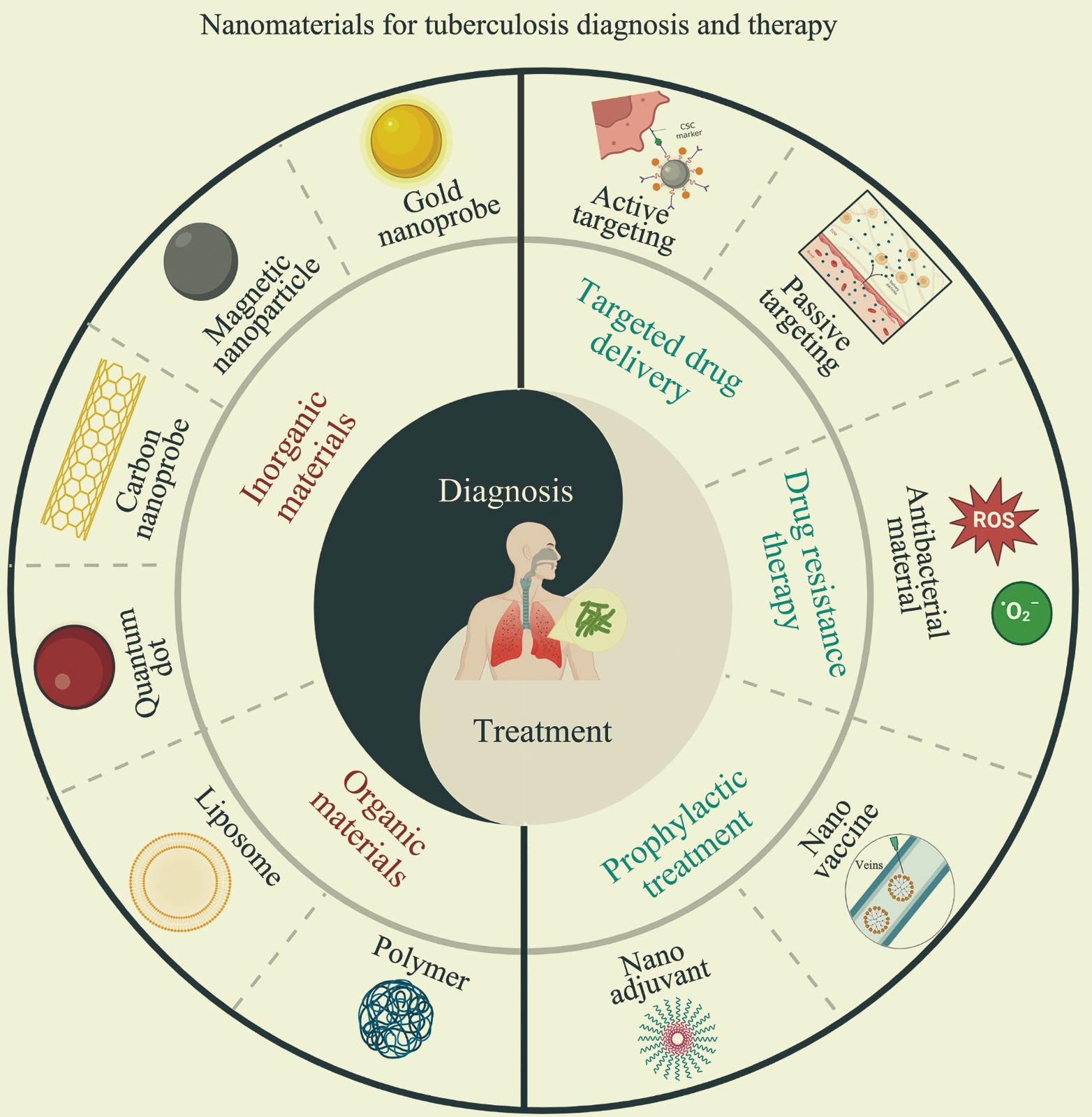
Application of Nanomaterials for Tuberculosis Diagnosis and Therapy
Effects of Zirconium Dioxide Nanoparticles Exposure on Histone H3 Modification in Human Skin Keratinocytes
The Therapeutic Potential of Extracellular Vesicles in Knee Osteoarthritis
Weighted Frequency-difference Electrical Impedance Tomography of Lung Based on RBFNN
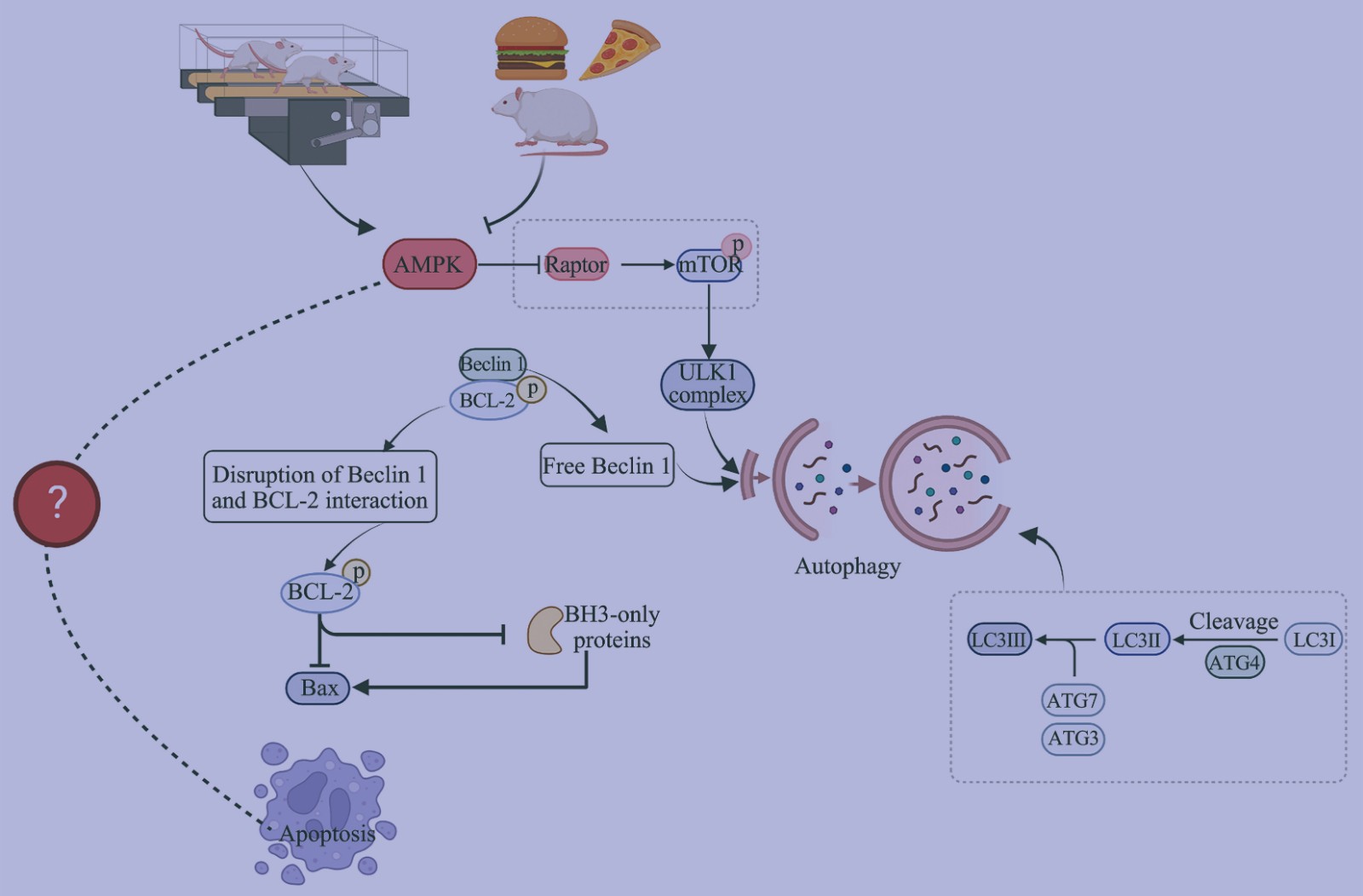
Effects of Exercise on Autophagy and Apoptosis in Testis Tissue of Obese Rats
Application of CRISPR/Cas9 Technology in Industrial Microorganisms
Sequestration of Cellular Transcription Factors by PolyQ-expanded Proteins and Its Effects on Gene Transcription Regulation
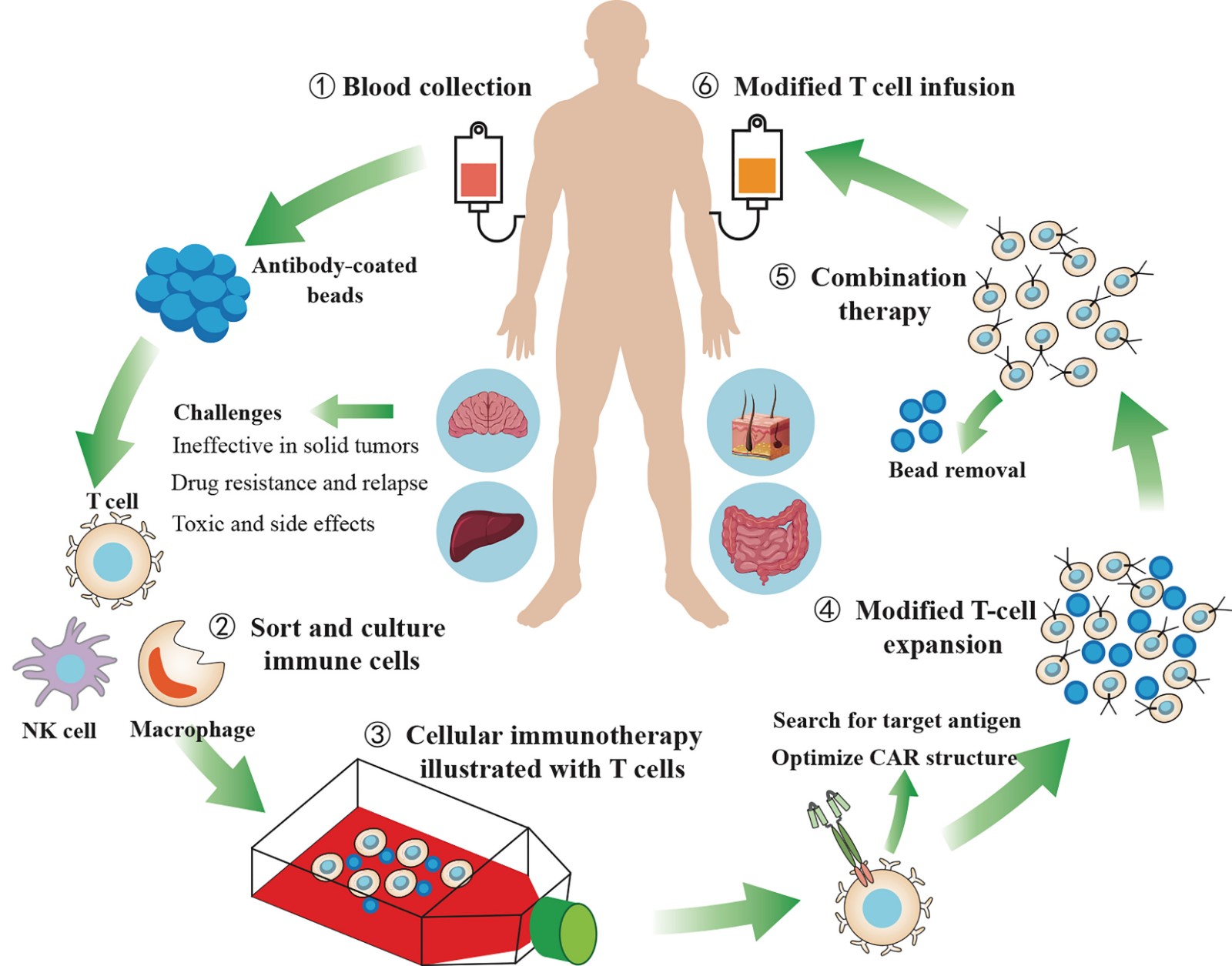
Current Status and Prospects of CAR-T Cell Therapy
Insight Into The Multi-faceted Roles of The Structure and Function of an Atypical Adenylate Kinase AK6/hCINAP
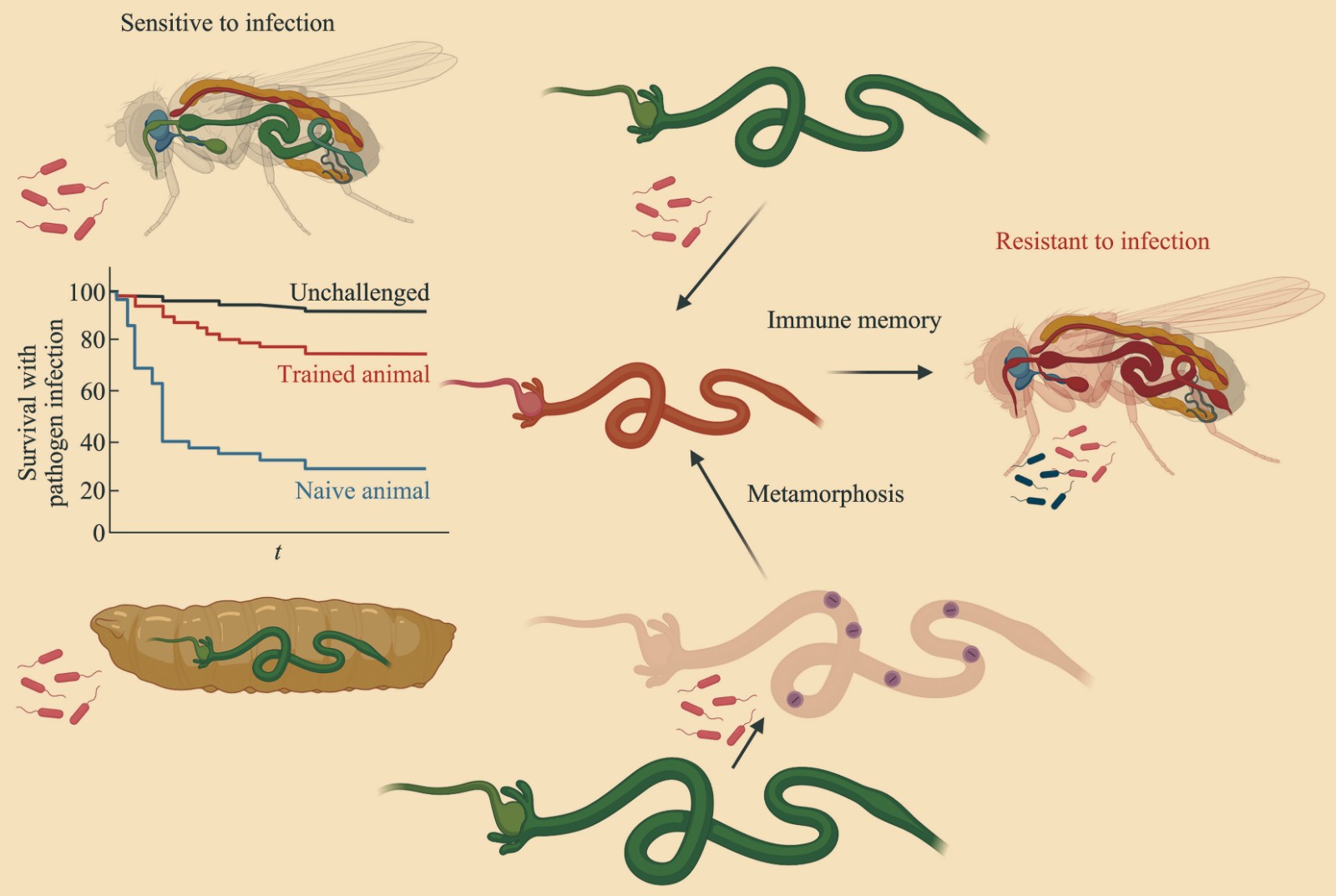
An Intestinal Trained Immunity Model Based on Drosophila melanogaster Oral Infection
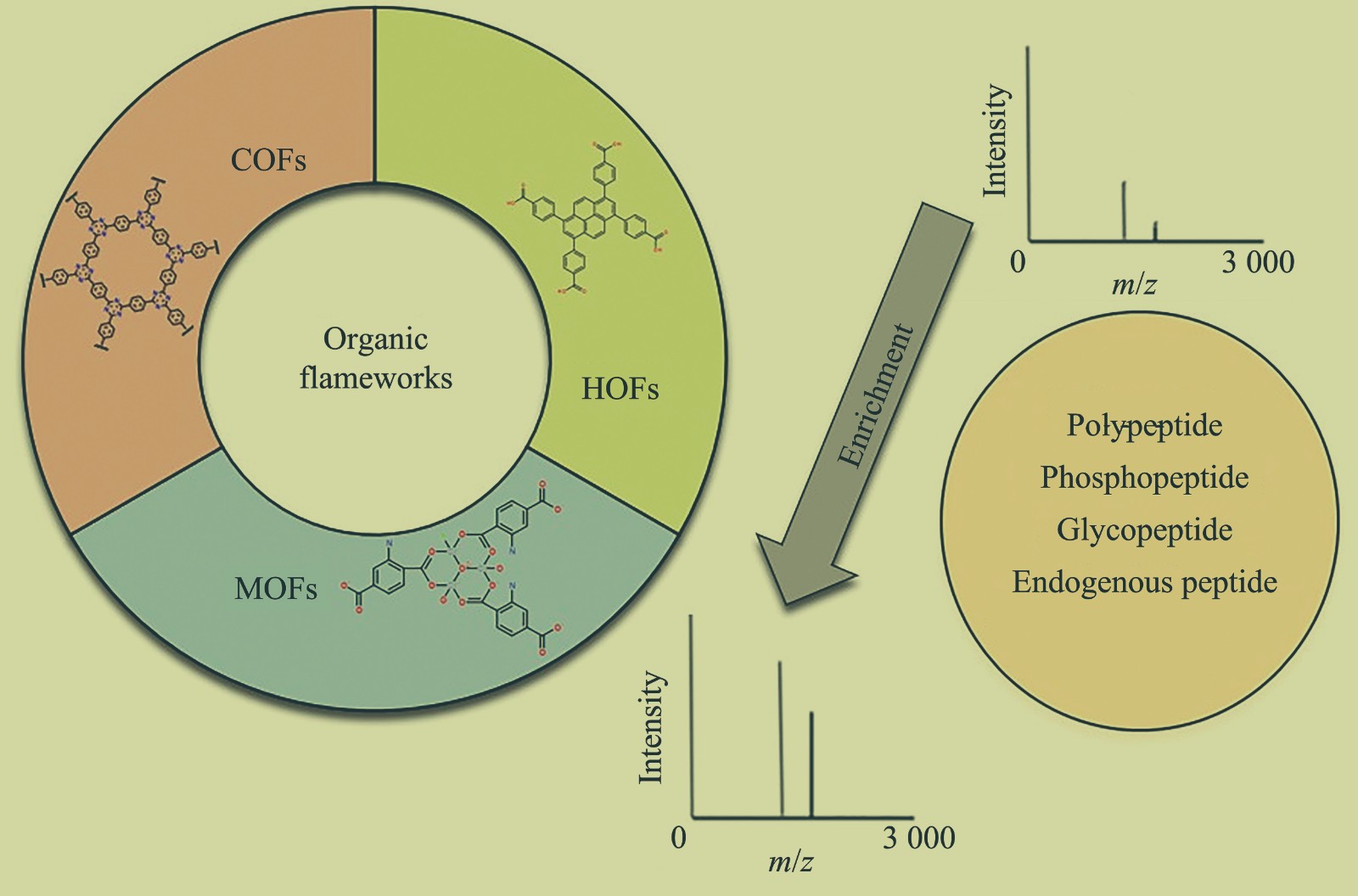
The Application of Organic Framework Materials for Peptide Enrichment
Predicting Synergistic Effect of Anticancer Drug Combinations Based on Nuclear Norm Regularization
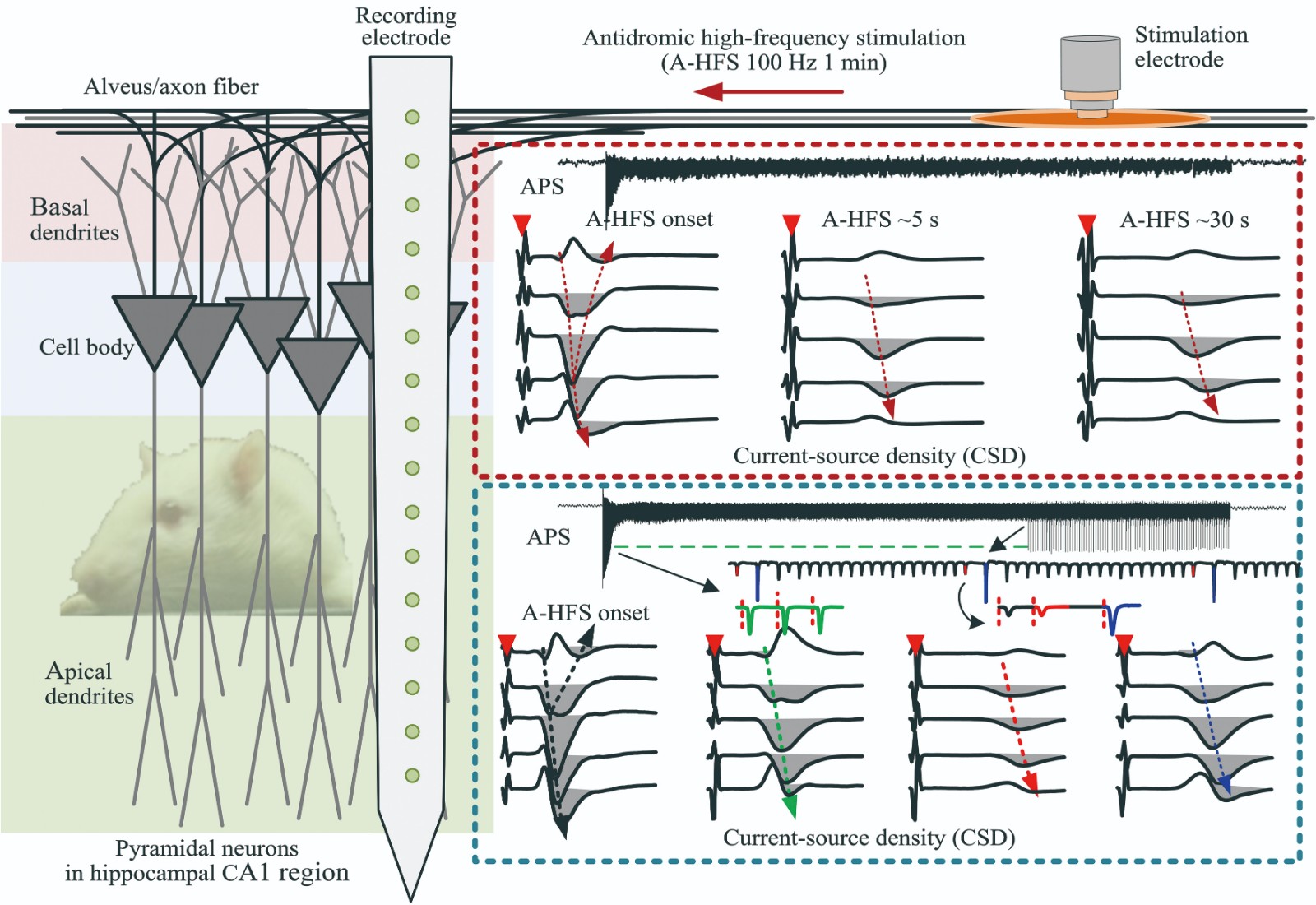
Effects on Somata by High-frequency Electrical Stimulation at The Axons of Hippocampal Pyramidal Neurons
Prediction Method of RNA Contact Map Based on Attention Mechanism